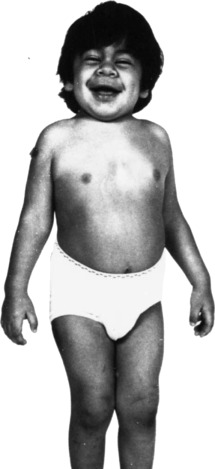
يتضمن داء عديد السكاريد المخاطي (MPS) نشاطًا معيبًا للإنزيمات الليزوزومية التي تؤدي إلى تحلل عديدات السكاريد المخاطية (الجليكوزامينوجليكان [GAGs] المرتبطة ببروتين رابط مع قلب حمض الهيالورونيك) إلى مكونات أصغر. تؤدي عملية التحلل غير المكتملة الناتجة إلى تراكم غير طبيعي لكبريتات الهيباران وكبريتات الديرماتان وكبريتات الكيراتان، ويتداخل التراكم غير الطبيعي لهذه المركبات مع وظيفة الخلية.
تم وصف أشكال مختلفة من MPS بشكل منفصل طوال القرن العشرين. تختلف أعراضها السريرية اعتمادًا على نوع الخلل الإنزيمي والبروتين السكري المتراكم.
أنواع داء عديد السكاريد المخاطي:
- متلازمة هيرلر (MPS IH).
- متلازمة هيرلر-شي (MPS I-H/S).
- متلازمة شي (MPS IS).
- متلازمة هنتر (MPS II).
- متلازمة سانفيليبو (MPS III).
- متلازمة موركيو (MPS IV).
- متلازمة ماروتو لامي (MPS VI).
- متلازمة ماكر (MPS السابع).
- متلازمة MPS IX وهو شكل نادر للغاية يتعلق بنقص الهيالورونيداز.
- متلازمة MPS X الناجم عن نقص انزيم الأريل سلفاتاز.
مسببات مرض داء عديد السكاريد المخاطي:
تنجم جميع اضطرابات MPS عن نقص أو خلل في إنزيم ليسوسومي محدد ضروري لتكسير كبريتات الديرماتان، أو كبريتات الهيباران، أو كبريتات الكيراتان، إما بمفردها أو معًا. يؤدي الفشل في تفكيك هذه السكريات المخاطية إلى تراكمها في الخلايا والأنسجة والأعضاء في جميع أنحاء الجسم. جميع هذه الاضطرابات موروثة كصفات جسمية متنحية باستثناء متلازمة هنتر، وهي سمة متنحية مرتبطة بالصبغي X.
يتم تحديد الأمراض الوراثية عن طريق التغيرات غير الطبيعية في جين الإنزيم الخاص بكل اضطراب، واحد من الأب والآخر من الأم. تحدث الاضطرابات الوراثية المتنحية عندما يرث الفرد جينًا غير طبيعي لنفس السمة من كل من الوالدين. إذا تلقى الفرد جينًا طبيعيًا واحدًا وجينًا واحدًا للمرض، فسيكون الشخص حاملًا للمرض، ولكن عادةً لن تظهر عليه الأعراض. إن خطر نقل الجين المعيب إلى الوالدين الناقلين، وبالتالي إنجاب طفل مصاب، هو 25٪ مع كل حمل. تبلغ نسبة خطر إنجاب طفل حامل للمرض مثل الوالدين 50% مع كل حمل. إن فرصة حصول الطفل على جينات طبيعية من كلا الوالدين وأن يكون طبيعيًا وراثيًا لهذه السمة المحددة هي 25٪.
الاضطرابات الوراثية المتنحية المرتبطة بالكروموسوم X هي حالات ناجمة عن جين غير طبيعي على الكروموسوم X. لدى الإناث اثنين من الكروموسومات X ولكن أحد الكروموسومات X “متوقف” ويتم تعطيل جميع الجينات الموجودة على هذا الكروموسوم. الأنثى التي لديها جين غير طبيعي موجود على أحد كروموسوماتها X هي حاملة لهذا الاضطراب. عادةً لا تظهر على الإناث الحاملة للمرض أعراض الاضطراب لأنه عادةً ما يتم إيقاف تشغيل الكروموسوم X الذي يحتوي على الجين غير الطبيعي. لدى الذكور كروموسوم X واحد، وإذا ورثوا كروموسوم X الذي يحتوي على جينة المرض، فسوف يصابون بالمرض. ينقل الذكور المصابون باضطرابات مرتبطة بالصبغي X جين المرض إلى جميع بناتهم، الذين سيكونون حاملين للمرض. لا يستطيع الذكور نقل الجين المرتبط بـ X إلى أبنائهم لأن الذكور ينقلون دائمًا كروموسوم Y بدلاً من كروموسوم X إلى ذريتهم الذكور. لدى الإناث الحاملات للاضطراب المرتبط بالصبغي X فرصة بنسبة 25% مع كل حمل لإنجاب ابنة حاملة مثلهن، وفرصة بنسبة 25% لإنجاب ابنة غير حاملة للمرض، و25% لإنجاب ابن مصاب بالمرض، و25% لإنجاب ابن مصاب بالمرض. فرصة 25% لإنجاب ابن غير متأثر.
النسبة المصابة بالمرض:
ويقدر معدل انتشار جميع أشكال داء عديد السكاريد المخاطي بنسبة واحدة من كل 25000 ولادة. ومع ذلك، نظرًا لأن عديدات السكاريد المخاطية، وخاصة الأشكال الخفيفة من الأمراض، غالبًا ما لا يتم التعرف عليها، فإن هذه الاضطرابات يتم تشخيصها بشكل ناقص أو يتم تشخيصها بشكل خاطئ، مما يجعل من الصعب تحديد معدل تكرارها الحقيقي في عموم السكان.
تتراوح تقديرات الأنواع المحددة من داء عديد السكاريد المخاطي من: واحد من كل 100000 لمتلازمة هيرلر؛ واحد من كل 500000 مصاب بمتلازمة شي؛ وواحد من كل 115000 مصاب بمتلازمة هيرلر-شي؛ واحد من كل 70.000 مصاب بمتلازمة سانفيليبو؛ واحد من كل 200000 مصاب بمتلازمة موركيو؛ وأقل من واحد من بين 250.000 في متلازمة سلاي. تحدث متلازمة هنتر في الغالب عند الذكور. وفي حالات نادرة للغاية، تم الإبلاغ عن إصابة الإناث. تقدر نسبة الإصابة بمتلازمة هنتر بواحد من كل 100.000 إلى 150.000 مولود ذكر.
تم تحديد أكثر من 40 مرضًا مختلفًا للتخزين الليزوزومي.
الأعراض والعلامات المرضية:
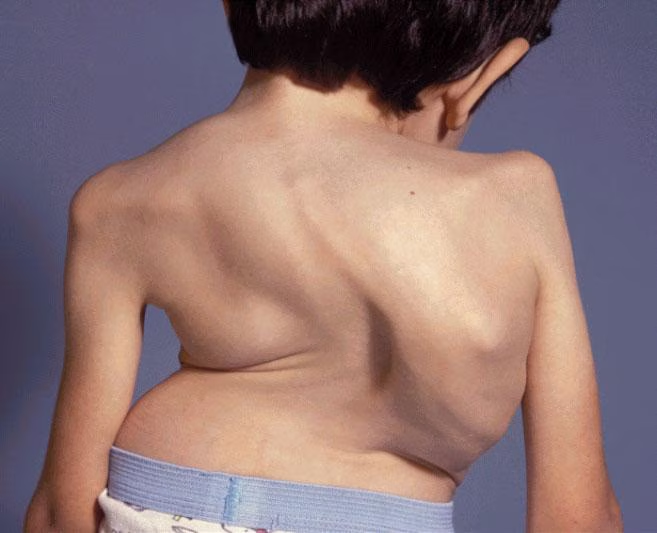
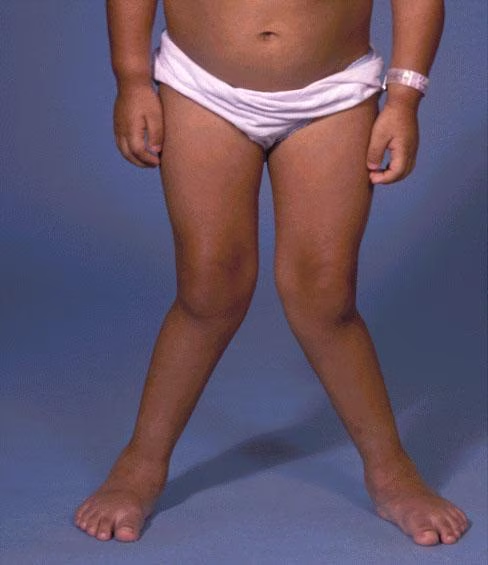
يتمتع المرضى الذين يعانون من MPS بنمو طبيعي في البداية، مع ظهور تشوهات في مرحلة الطفولة أو في وقت لاحق في مرحلة الطفولة. قد يكون لدى الأشخاص الذين يعانون من مشاكل عديدة في بعض الأجهزة التالية:
- أمراض الجهاز العصبي المركزي – استسقاء الرأس. اعتلال النخاع الشوكي العنقي.
- أمراض القلب والأوعية الدموية – الذبحة الصدرية. ضعف الصمامات. ارتفاع ضغط الدم. قصور القلب الاحتقاني.
- أمراض رئوي – انسداد مجرى الهواء، مما قد يؤدي إلى انقطاع التنفس أثناء النوم، أو قصور شديد في الجهاز التنفسي، أو القلب الرئوي.
- أمراض العيون – غشاوة القرنية. الجلوكوما. وذمة حليمية مزمنة. تنكس الشبكية.
- ضعف السمع – الصمم.
- أمراض العضلات والعظام – قصر القامة. تصلب المفاصل أعراض انحباس العصب المحيطي.
تشخيص داء عديد السكاريد المخاطي:
تتضمن دراسات الفحص السابقة للولادة التي قد تكون مفيدة ما يلي:
- أخذ عينات من الزغابات المشيمية.
- بزل السلى.
تشمل الدراسات التشخيصية بعد الولادة التي قد تكون مفيدة ما يلي:
- تحليل البول، مع التركيز على الجليكوسامينوجليكان (على سبيل المثال، كبريتات ديرماتان، وكبريتات الهيبارين، وكبريتات الكيراتين).
- فحوصات المصل للإنزيمات الليزوزومية (alpha-L-iduronidase، iduronate sulfatase، heparan N-sulfatase، N-acetylglucosaminidase، alpha-glucosamine- N-acetyltransferase، N-acetyl alpha-glucosamine-6-sulfatase، N-acetylgalactosamine-6-sulfatase , (ن-أسيتيل جالاكتوزامين-6-سولفاتيز، ب-جالاكتوزيداز، ن-أسيتيل جالاكتوسامين-4-سولفاتيز، و ب-جلوكورونيداز).
دراسات التصوير التي قد يكون لها ما يبررها هي كما يلي:
- التصوير الشعاعي العادي (للكشف عن خلل التعظم المتعدد).
- التصوير المقطعي المحوسب (CT) للجمجمة (للمساعدة في تشخيص استسقاء الرأس).
- تخطيط صدى القلب (لمراقبة وظيفة البطين وحجمه لدى مرضى MPS المصابين بأمراض القلب والأوعية الدموية).
- التصوير بالرنين المغناطيسي (MRI؛ لتقييم إصابة العمود الفقري).
الاختبارات الأخرى التي يجب مراعاتها هي كما يلي:
- تخطيط كهربية الشبكية.
- التقييم السمعي.
العلاجات الدوائية وغير الدوائية:
للأسف ان العلاجات محدودة بالنسبة لالداء المذكور. وقد اقتصرت على الرعاية الداعمة وطرائق العلاج التجريبية. تشمل طرق العلاج الطبي ما يلي:
- لارونيداز ( يحفز التحلل المائي لمخلفات حمض ألفا-إل-إيدورونيك الطرفية من كبريتات الديرماتان وكبريتات الهيبارين).
- إدورسولفاس (إنزيم الليزوزوم المنقى يستخدم كعلاج بديل لمتلازمة هنتر لدى المرضى من الأطفال والبالغين).
- إلوسولفاز ألفا (يُشار إلى إنزيم الجليكوسامينوجليكان الليزوزومي (GAG) كعلاج بديل للإنزيم لداء عديد السكاريد المخاطي من النوع IV A).
- فيسترونيداز ألفا (العلاج ببدائل الإنزيم لعلاج داء عديد السكاريد المخاطي من النوع السابع (MPS VII)).
قد تشمل الرعاية الجراحية لحالات معينة ما يلي:
- استسقاء الرأس – تحويلة البطين الصفاقي.
- عتامة القرنية – زراعة القرنية.
- أمراض القلب والأوعية الدموية – استبدال الصمامات.
- مرض انسداد مجرى الهواء – ثقب القصبة الهوائية.
- حالات العظام – تحرير النفق الرسغي؛ إجراءات الأنسجة الرخوة لتحرير تقلصات الورك والركبة والكاحل؛ جراحات احتواء الورك؛ قطع العظم التصحيحي لتشوه الأروح التدريجي في الركبة؛ الانصهار الفقري الخلفي.
تعد الرعاية متعددة التخصصات إلزامية لهؤلاء المرضى ويجب أن تشمل طبيب أطفال (طبيب باطني)، وطبيب أعصاب، وطبيب قلب، وطبيب عيون، وأخصائي سمع، وجراح عظام، ومعالج طبيعي ومهني.
المراجع:
- https://emedicine.medscape.com/article/1258678-overview?&icd=login_success_email_match_fpf
- https://go.drugbank.com/drugs/DB01271
- https://go.drugbank.com/drugs/DB00090
- https://go.drugbank.com/drugs/DB09051
- https://reference.medscape.com/drug/vimizim-elosulfase-alfa-999909
- https://go.drugbank.com/drugs/DB12366
- https://rarediseases.org/rare-diseases/mucopolysaccharidoses/

GIPHY App Key not set. Please check settings